What is Spinal Muscular Atrophy (SMA)?
Reviewed by: HU Medical Review Board | Last reviewed: August 2021 | Last updated: October 2025
Spinal muscular atrophy (SMA) is a genetic disorder where muscles become weak and waste away. It happens when spinal cord nerve cells die, causing important muscles to become inactive. It is the most common genetic cause of death in infants.1,2
Symptoms and outcomes vary depending on the type of SMA. The most common and severe type is type 1, where symptoms begin during the first few months of life. Milder types affect older children and adults in different ways.1,2
There is still no cure for SMA, but new treatments are improving health outcomes. Many possible treatments are being researched or are being studied in clinical trials.
What causes spinal muscular atrophy?
In most cases, SMA is caused by low levels of a protein called the “survival of motor neuron” (SMN) protein. The SMN protein is made with instructions from 2 regions of our DNA, called the SMN1 gene and the SMN2 gene. People usually have 2 copies of each gene, with 1 from each parent. The SMN1 genes provide the instructions for most of the SMN protein produced by our cells.1,2
In nearly all people with SMA, the SMN1 gene is altered and cannot be used to produce any SMN protein. SMA occurs when a child inherits 2 copies of the altered SMN1 gene, with 1 from each parent.3
You can imagine the SMN1 gene as a recipe that our cells have to follow to make the SMN protein. Our cells have 2 copies of the recipe and can use the good 1 in case 1 has a mistake. However, in SMA there is a mistake in both copies of the recipe. This causes the protein prepared by cells to be non-functional.
The lack of SMN protein causes motor neurons in the spine to gradually die. Motor neurons are cells that control muscle movements needed for speaking, walking, and breathing. When motor neurons in the spine die, signals starting in the brain cannot travel to the muscles. These muscles weaken and waste away, affecting essential movements.1,4
Figure 1. SMA-affected nerve and muscle cells lead to atrophied muscles
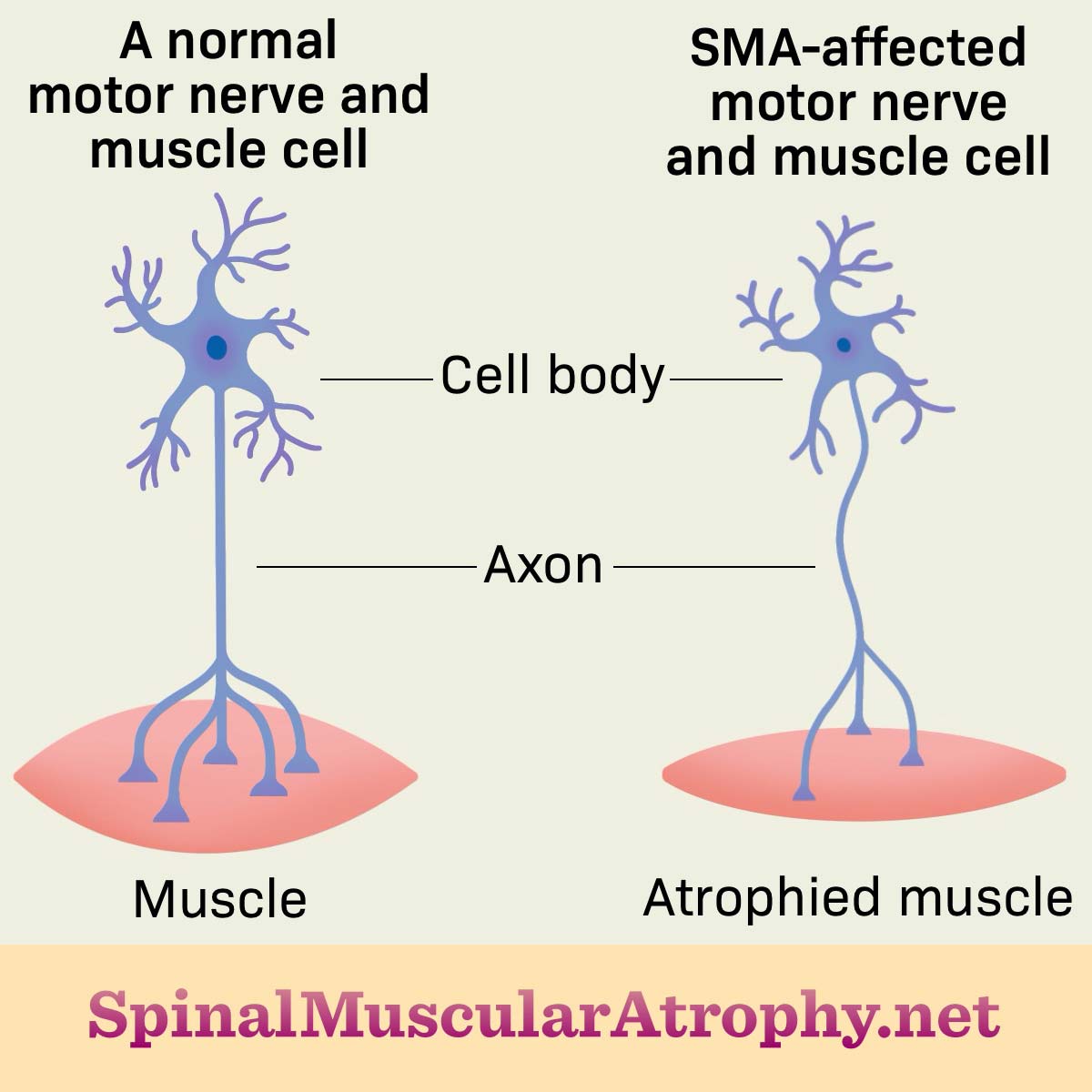
What are the different types?
SMA is actually a group of disorders that vary in severity. The severity depends mostly on how well the SMN2 genes can make enough SMN protein in the absence of SMN1. Most people have 2 copies of the SMN2 gene, but having more than 2 copies usually means the disease will be milder.2,5
SMA is divided into 4 major types. The types are defined based on the highest motor development milestone a person reaches:6,7
- Type 1 – Also called Werdnig-Hoffmann disease, this is the most severe and common type of SMA. Symptoms begin within 6 months of life, and infants can never sit. The disease progresses quickly, and infants usually die by age 2 from respiratory infections.
- Type 2 – Also called Dubowitz disease, this is an intermediate form of SMA. Symptoms appear between 7 and 18 months old, and most children with this type can never stand. Life expectancy ranges from early childhood to adulthood.
- Type 3 – Also called Kugelberg-Welander, this is a mild form of SMA. Symptoms appear from 18 months old to early adulthood. People can stand and walk, but they may have muscle weakness. Life expectancy is normal.
- Type 4 – This is an adult form of SMA. Symptoms appear after age 30. People usually experience slowly progressive muscle weakness and other symptoms.
Newer classifications are also being used. For example, some experts classify people as “sitters” or “walkers” based on their functional status.8-10
How common is this condition?
About 1 in 6,000 to 10,000 babies worldwide are born with SMA. More than 25,000 people in the United States have SMA. People of every race and gender are equally affected by SMA.2
About 6 million people in the United States are carriers of the SMA gene. This is roughly 1 in 50 people. This means they have 1 copy of a normal SMN1 gene and 1 copy of the mutated version. They can pass on the gene to their biological children but are otherwise healthy.6,11
What are the symptoms and complications of SMA?
Symptoms of SMA vary depending on the type, stage, and other factors specific to each person. The most common early symptoms of type 1 SMA are floppy limbs and poor head control. Other common symptoms for type 1 include:2,7
- Reduced muscle tone
- Low limb movements and reflexes
- Swallowing, feeding, and breathing difficulties
- Muscle twitches
- Skeletal abnormalities
- Difficulty sitting
Symptoms for other types of SMA tend to be milder and start later in life.
What are the health outcomes and how do they vary?
Health outcomes for people with SMA vary depending on the type. The most severe forms (types 1 and 2) are fatal, usually because of respiratory failure. This develops when the lungs cannot get enough oxygen into the blood. Children with SMA have normal brain development and are highly intelligent and social.2
Most people with type 3 or 4 have a normal life expectancy. However, muscle weakness and other symptoms can still interfere with daily life.3
There is no cure for SMA. Treatment is focused on reducing symptoms and preventing complications. The FDA has approved 3 treatments for SMA. These have changed the course of SMA progression for many people. These treatments are:6,8-10
- Spinraza® (nusinersen)
- Zolgensma® (onasemnogen abeparovec-xioi)
- Evrysdi™ (risdiplam)
People with SMA are usually treated by a diverse team of doctors. Treatment from these specialists may include:8-10
- Physical therapy and occupational therapy
- Speech therapy
- Assistive devices
- Nutritional support
- Respiratory (breathing) support